Mutation Query
| | | | Allele 1: | A862T | | Allele 2: | H277L | Allelic information known | | Refine query |
|
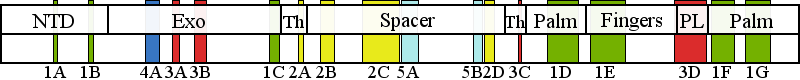
| | | Residue H277 | | Cluster assignment: | | | Cluster description: | Partitioning loop | | Subcluster: | 3A (residues 268-277) | | Subcluster description: | A region near the exo active site that comprises the Exo II motif | | POLG domain: | Exonuclease domain |
| Residue A862 | | Cluster assignment: | | | Cluster description: | Polymerase active site and environs | | Subcluster: | 1D (residues 848-895) | | Subcluster description: | This subcluster forms a large portion of the pol active site and contains two highly conserved motifs that are found in all family A polymerases: the RR loop (motif 2) and motif A (Loh and Loeb, 2005). | | POLG domain: | Polymerase domain |
|
|
Mutation Information
|
| A862T | | | | Number of patients: (with A862T) | 7 | | Found together with: | |  | Show Patient Data |
| Patient data are sorted by mutation combination frequency. | Reference: | Stricker et al, 2009; | | Description: | Progressive cerebellar ataxia, neuropathy, restless legs syndrome, hemihypesthia, myoclonic epileptic seizures, severe ataxia, dysphagia, muscle strength preserved, migraines, headaches, abdominal pain, death via prolonged status epilepticus. | | Mutations: | A862T, R964C | | Age group: | juvenile | | Age of Onset: 15, Age of Patient: n/a, Age of Death: 27 |
| Reference: | Stricker et al, 2009; | | Description: | Progressively focal motor and tonic-clonic seizures, delayed psychomotor development, sensoriaxonal neuropathy, mild tetraparesis, cerebellar syndrome, intestinal pseudoobstruction, died via refractory status epilepticus. | | Mutations: | A862T, R964C | | Age group: | childhood | | Age of Onset: 5, Age of Patient: n/a, Age of Death: 23 |
| Reference: | Wong et al, 2008; | | Description: | Onset 17 years with ataxia, exercise intolerance, cerbellar atrophy, SCAE, seizures, dementia. | | Mutations: | A862T, R964C | | Age group: | juvenile | | Age of Onset: 17, Age of Patient: n/a, Age of Death: n/a |
| Reference: | Stewart et al, 2009; | | Description: | PEO with ataxia, SCA-like phenotype in siblings, multiple deletions in muscle mtDNA detected via LPCR. 3% COX deficient fibers. | | Mutations: | A862T, R1047W | | Age group: | adult | | Age of Onset: 61, Age of Patient: n/a, Age of Death: n/a |
Back to top | Reference: | Lax et al, 2012a; | | Description: | CPEO, Ptosis, Peripheral neuropathy, COX-deficient fibers, presence of mitochondrial dna deletions in muscle, ataxia, dysarthria, Axonal sensory neuropathy /neuronopathy, Distal and proximal neurogenic change. | | Mutations: | A862T, R1047W | | Age group: | adult | | Age of Onset: 22, Age of Patient: 61, Age of Death: n/a |
| Reference: | Ferreira et al, 2011; | | Description: | Onset at 2.5 years with epilepsy, diagnosed as Alpers. Complex IV 33%. showed psychomotor regression around age 2.5 years, and developed repetitive generalized tonic–clonic seizures. Myoclonic jerks. | | Mutations: | A862T, R1081Q | | Age group: | childhood | | Age of Onset: 3, Age of Patient: 7, Age of Death: n/a |
| Reference: | McKelvie et al, 2012; | | Description: | Ataxia, sensory ataxic neuropathy, with cerebellar features, visual disturbances with diplopia, dysarthria and dysphasia. Multiple mtDNA seletions, COX-negative fibers and ragged red fibers were found in autopsy. Areflexic, absent reflexes, in all limbs, distal weakness and distal sensory loss of proprioception and vibration. She became encephalopathic and febrile. | | Mutations: | A862T, H277L | | Age group: | adult | | Age of Onset: 46, Age of Patient: 54, Age of Death: 66 |
|
| H277L | | | | Number of patients: (with H277L) | 5 | | Found together with: | | | Show Patient Data |
| Patient data are sorted by mutation combination frequency. | Reference: | Hunter et al, 2011; | | Description: | Developmental Delay or Regression, hypotonia, vomiting, Abnormal Liver Enzymes, liver mtDNA depletion, clinical diagnosis of infantile hepatopathy | | Mutations: | A467T, H277L, R232H | | Age group: | infantile | | Age of Onset: 0.125, Age of Patient: n/a, Age of Death: 0.25 |
| Reference: | Hunter et al, 2011; | | Description: | Developmental Delay or Regression, motor paresis, hypotonia, vomiting, Abnormal Liver Enzymes, Serum Lactate, liver mtDNA depletion, clinical diagnosis of infantile hepatopathy | | Mutations: | A467T, H277L, R232H | | Age group: | infantile | | Age of Onset: 0.17, Age of Patient: n/a, Age of Death: 0.25 |
| Reference: | Ashley et al, 2008; | | Description: | Onset at 6 months of age, epilepsy, hepatopathy. 4% mtDNA copy number in liver, 32% mtDNA copy number in muscle. | | Mutations: | H277L, T851A | | Age group: | infantile | | Age of Onset: 0.5, Age of Patient: n/a, Age of Death: n/a |
| Reference: | Sato et al, 2011; | | Description: | left dominant hemi-parkinsonian features, bradykinesia, gait disturbance, resting tremor and postural instability, chronic progressive external ophthalomoplegia among mitochondrial myopathies | | Mutations: | H277L, R943C | | Age group: | adult | | Age of Onset: 50, Age of Patient: 78, Age of Death: n/a |
Back to top | Reference: | McKelvie et al, 2012; | | Description: | Ataxia, sensory ataxic neuropathy, with cerebellar features, visual disturbances with diplopia, dysarthria and dysphasia. Multiple mtDNA seletions, COX-negative fibers and ragged red fibers were found in autopsy. Areflexic, absent reflexes, in all limbs, distal weakness and distal sensory loss of proprioception and vibration. She became encephalopathic and febrile. | | Mutations: | A862T, H277L | | Age group: | adult | | Age of Onset: 46, Age of Patient: 54, Age of Death: 66 |
|
|
|
|
|
The following information is based on existing patient data and pathogenic cluster assignment.
Pathogenicity information for a patient with mutations in Clusters 1 and 3: Age of onset information is extracted from a total of 18 patients and/ or patient families. | Age of onset | | |
18- 9- | 9
| 2
| 2
| 5
| | | infantile | childhd | juvenile | adult | | | 50% | 11% | 11% | 28% | |
All mutations mapping within the pathogenic clusters are at high risk for pathogenicity. In general, a patient must have a pathogenic mutation in both of his/ her POLG genes to develop a POLG-related syndrome. | Symptoms described in patients with cluster1-cluster3 mutations | |
| Symptoms in patients with combination
cluster1:cluster3 | | Movement disorder (ataxia) | 44.4% | | Encephalopathy | 33.3% | | Developmental delay | 33.3% | | Epilepsy | 27.8% | | PEO | 27.8% | | Failure to thrive | 22.2% | | Hypotonic | 22.2% | | Peripheral neuropathy | 16.7% | | Ptosis | 16.7% | | Dysarthria | 16.7% | | Lactic acidosis | 11.1% | | Ragged red fibers | 11.1% | | Muscle weakness | 11.1% | | Liver failure | 11.1% | | Headache/ migraine | 11.1% | | Dementia | 11.1% | | Retardation | 11.1% | | GI dysmotility | 11.1% | | Myoclonic seizures | 5.6% | | Sensory ataxia | 5.6% | | Polyneuropathy | 5.6% | | Demyelinating neuropathy | 5.6% | | Axonal sensorimotor polyneuropathy | 5.6% | | Abnormal muscle ultrastructure | 5.6% | | Exercise intolerance | 5.6% | | Cox-negative | 5.6% | | Diplopia | 5.6% | | Liver dysfunction | 5.6% | | Growth retardation | 5.6% | | Vomiting | 5.6% | | GI reflux | 5.6% | | Cyclic vomiting | 5.6% | | Delayed gastric emptying | 5.6% | | Tremor | 5.6% | | Hearing loss | 5.6% |
| | Data gathered from clinical descriptions for 18 patients |
| Symptoms by group | | Developmental Delay | 50.0% | | Ataxia | 44.4% | | CPEO | 38.9% | | Seizures | 33.3% | | Neuropathy | 27.8% | | Alpers syndrome | 22.2% | | CNS symptoms | 22.2% | | Hypotonia | 22.2% | | GI symptoms | 16.7% | | Hepatopathy | 16.7% | | Myopathy | 16.7% | | Migraines | 11.1% | | Other | 5.6% |
| | [Show grouping information] |
|
|
|